CRISPR-Cas
CRISPR-Cas is user-friendly, affordable, and can target multiple genome sites simultaneously, making it a highly successful genome editing tool.
Introduction
Over the past few years, genome editing techniques have experienced an impressive development and have been finely tuned to meet researchers’ needs in terms of accuracy, time consumption, and costs. Since the first experiments elucidating the utility of restriction enzymes in the early 1970s, the use of restriction endonucleases has become a common tool in research labs worldwide (1).
One of the striking advantages of using endonucleases for genome engineering became obvious with the finding that genomic DNA undergoes recombination events following double strand breaks (DSBs), both in yeast and in mammalian cells (2; 3; 4). One approach aimed to genome engineering was targeting restriction enzymes specifically to different genome loci by modifying the enzyme recognition site (4). The main disadvantage of such an approach is that both, DNA recognition site and DNA cleavage site are present in the same endonuclease domain (5). Therefore, modifying the DNA recognition site of an endonuclease can result in the alteration of the nuclease protein structure itself (4).
So far, the most successful approach to genome engineering is represented by using systems where a DNA cutting moiety is complemented with a separate DNA recognition moiety. In Zinc finger nucleases and Transcription activator-like effector nuclease (TALEN), the DNA cutting and the DNA recognition moieties are represented by different domains of an engineered endonuclease (6). Alternatively, in the Clustered regularly interspaced short palindromic repeats (CRISPR)- CRISPR associated protein (Cas) (CRISP-Cas) system, the DNA recognition moiety consists of an RNA guide sequence that forms a ribonucleoprotein complex with a Cas nuclease (6).
Compared to other genome editing techniques, CRISPR-Cas is relatively easy to use, it is cost-effective and importantly, it offers the opportunity to target multiple genome sites in parallel. These unique features have made so far the success of this new genome editing tool (6).
CRISPR-Cas systems in nature
CRISPR-Cas are a natural immunity mechanism developed by bacteria and archaea to neutralize invading DNA coming from viruses or plasmids (7). This system acts in three main stages (Figure 1). In the first stage, the invading DNA is incorporated in the host genome in fragments (protospacers), interspaced between CRISP repeats (6; 8). In a second stage, CRISP repeats and protospacers are processed into small CRISPR RNA (crRNA). In the third and last stage, sequence-specific crRNAs associate with Cas nucleases and form a ribonucleoprotein complex, which recognizes and destroys the invading nucleic acid (6; 8).

Figure 1. CRISPR-Cas system is organized in three stages. First, the invading DNA is incorporated into the host genome in the form of a protospacers, interspaced between CRISP repeats. In the second stage, the crRNA is expressed. In the third stage, the ribonucleoprotein complex formed by crRNA and Cas nuclease(s) cleaves and destroys the invading DNA (modified from 17).
CRISPR RNAs interact with Cas nucleases in different ways, depending on the CRISPR-Cas system.
Three main types of CRISPR-Cas systems are observed in nature. In type I and III, pre-crRNAs are processed by dedicated Cas nucleases into mature crRNAs, each of which assembles with Cas nucleases into a multi-Cas protein complex that targets the invading DNA. In type II, each mature crRNA assembles into a two-RNA complex with a transactivating-RNA (tracr-RNA). This dual-RNA complex then binds a Cas nuclease to form a ribonucleoprotein complex, which cleaves the invading DNA (8). CRISPR-Cas type II has been extensively studied and it was implemented in 2012 as a novel biotechnological tool aimed to genome editing (8).

Figure 2. CRISPR-Cas type II. (a) Protospacers and CRISPR repeats are processed into small CRISPR-Cas RNA (crRNA). Each crRNA hybridizes to a tracrRNA and form a complex with the Cas9 nuclease. This complex recognizes and cleaves foreign DNAs bearing the protospacer sequences. (b) The CRISPR-Cas system utilizes a fusion between a crRNA and part of the tracrRNA sequence. This single gRNA complexes with Cas9 to mediate cleavage of target DNA sites (6).
In CRISPR-Cas type II, crRNAs typically have two sequence domains, one homologous to the target protospacer sequence and one homologous to a transactivating CRISPR RNA (tracrRNA; Figure 2a). This latter domain allows each crRNA to hybridize with a tracrRNA, before binding the Cas 9 nuclease. The association of Cas9, crRNA, and tracrRNA results in the formation of a ribonucleoprotein complex that triggers, cuts and inactivates the invading DNA (9). For a faithful cut, the target DNA must bear a protospacer sequence at the immediate 5’ of a short protospacer adjacent motif (PAM; Figure 3). For S. pyogenes Cas 9, PAM corresponds to an NGG motif (6). Cas nucleases derived from other bacterial strains recognize different PAMs; for example, Cas9 from S. aureus recognizes a NNGRRT PAM (10).
CRISPR-Cas in genome editing
Since 2012, the CRISPR system was implemented into a programmable dual-RNA-guided DNA endonuclease (8). Jinek and colleagues showed that CRISPR-Cas can be targeted to virtually any dsDNA sequence having a PAM at the immediate 3’-end of the DNA target (protospacer) sequence. This process triggers a DSBs in the target DNA (8).

Figure 3. CRISPR-Cas9 recognition site. In nature, a crRNA anneals to a complementary protospacer of the invading DNA, and hybridizes with a specific tracrRNA. These RNAs form then a ribonucleoprotein complex with Cas9 (8).
This 2 RNA (crRNA and tracrRNA) based system, was further developed into a single guide RNA (sgRNA) sequence of 48 bases in length, having 20 bases of DNA recognition site (Figure 2b; 8). However, the most commonly used version of sgRNA has a sequence of 85 bases (11; 12).
Both as a single sgRNA or a dual-RNA system, CRISPR-Cas has been now extensively used for genome editing in a variety of cell-types and organisms, including plants, mammals and human pluripotent stem cells in culture (6). This is due to the fact that DSBs activate a repair system in the host organism, which can repair the damage either by non-homologous end joining (NHEJ) or by homology directed repair (HDR). NHEJ usually results in insertions or deletions of DNA stretches in the target DNA sequence, which causes frameshift mutations and blocks the downstream protein translation. This kind of mutations has also been used to disrupt the binding sites of gene activators/silencers. HDR repair system can be used instead to insert donor-DNA sequences in specific points of the genome.
A critical point towards a successful CRISPR-Cas9 genome editing: designing the crRNA.
The protospacer or gRNA sequence:
Whether your final aim consists in disrupting or in modulating the protein synthesis of a target gene, designing your crRNA is a critical step towards a faithful genome editing. Particular attention should be given to the target protospacer sequence within your crRNA (gRNA; 13). In average, this sequence is 20 bases long and needs to be wisely chosen, to meet at least two important criteria:
- The gRNA sequence within the crRNA needs to complement to a target DNA sequence being in close proximity to a PAM (6; 13). In case you are aiming to use the Type II CRISPR system from S. pyogenes, PAM needs to sit at the immediate 3’-end of the gRNA target sequence (6). Note that the PAM site must be present on the target DNA but must not be included into your crRNA!
- The gRNA target sequence must recur only once in the targeted genome. Identical or highly-similar loci in the genome may lead to your Cas nuclease cutting an off-target site. Cas nucleases have a mismatch tolerance as high as 4-5 nucleotides towards the recognition of the protospacer site (14). Therefore, DNA sequences that differ 4-5 nucleotides from the intended target sequence can still be recognized and cleaved by Cas enzymes, which results into hundreds of potential off-target sites within the human genome (6).
There are a number of online tools to design gRNA sequences and minimize unwanted off-target effects in your CRISPR genome-engineering experiment. However, a major limitation of these tools is the small number of available genomes (13). A new online tool called Breaking-Cas was recently developed, which offers the possibility to analyze any eukaryotic genome present on ENSEMBL and ENSEMBLGENOMES databases (13). Moreover, this tool presents two important features.
First, it allows expert users to modify and customize both, sequence and location of PAM.
Second, users can customize the length of a gRNA sequence to be between 18 and 25 nucleotides. These features allow users to design genome editing experiments with Cas nucleases like FnCpf1, whose cleavage site is distant from the PAM sequence and cleavage occurs after the 18th base on the non-targeted strand and after the 23rd base on the targeted strand (15).
At metabion we are proud to recommend our customers this state-of-the-art CRISP-Cas9 design-tool developed at the Spanish National Centre for Biotechnology (CNB-CSIC). You can access it from our website.
Hybridization with tracrRNA
In nature, crRNA is 42 nucleotides long. Of these, 22 residues are involved in binding tracrRNA (11). However, the minimal binding region between crRNA and tracrRNA was shown to be as short as 10 nucleotides (8). Within this sequence, there is typically a stretch of 4 Ts, which can be detrimental for the actual transcription of your sgRNA, as a sequence of 4 Ts represents the termination sequence of RNA Polymerase III (11; 16). Further evidence has shown that the crRNA:tracrRNA binding can be improved by extending the homology region between crRNA and tracrRNA of about 5 base-pairs. This increases Cas9 activity and results in a dramatic improvement of knock-out efficiency (11; 12). Best knock-out efficiency was achieved upon additionally mutating the 4T-stretch within crRNA into TTTC (Figure 4; 11). Therefore, it is advisable to design crRNA having 20 nucleotides of homology to the target DNA and 15 basis of homology to the tracrRNA (11).
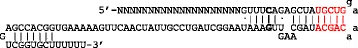
Figure 4. Optimization of sgRNA. The letters in bold highlight the mutation of the UUUU stretch into UUUC (11).
For our RNA longmers portfolio please click here.
References
- Roberts RJ. How restriction enzymes became the workhorses of molecular biology. Proc Natl Acad Sci U S A. 2005 Apr 26;102(17):5905-8.
- Rudin N., Sugarman E., Haber JE. Genetic and physical analysis of double-strand break repair and recombination in Saccharomyces cerevisiae. Genetics. 1989 Jul;122(3):519-34.
- Rouet P., Smih F., Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994 Dec;14(12):8096-106.
- Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011 Aug;188(4):773-82.
- Ashworth J., Havranek JJ., Duarte CM., Sussman D., Monnat RJ. Jr, Stoddard BL., Baker D. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006 Jun 1;441(7093):656-9.
- Sander JD., Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014 Apr;32(4):347-55.
- Wiedenheft B., Sternberg SH., Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012 Feb 15;482(7385):331-8.
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna JA., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21.
- Deltcheva E., Chylinski K., Sharma CM., Gonzales K., Chao Y., Pirzada ZA., Eckert MR., Vogel J., Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011 Mar 31;471(7340):602-7.
- Nishimasu H., Cong L., Yan WX., Ran FA., Zetsche B., Li Y., Kurabayashi A., Ishitani R., Zhang F., Nureki O. Crystal Structure of Staphylococcus aureus Cas9. Cell. 2015 Aug 27;162(5):1113-26.
- Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015 Dec 15;16:280.
- Chen B., Gilbert LA., Cimini BA., Schnitzbauer J., Zhang W., Li GW., Park J., Blackburn EH., Weissman JS., Qi LS., Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013 Dec 19;155(7):1479-91.
- Oliveros JC., Franch M., Tabas-Madrid D., San-León D., Montoliu L., Cubas P., Pazos F. Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016 Jul 8;44(W1):W267-71.
- Fu Y., Foden JA., Khayter C., Maeder ML., Reyon D., Joung JK., Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013 Sep;31(9):822-6.
- Zetsche B., Gootenberg JS., Abudayyeh OO., Slaymaker IM., Makarova KS., Essletzbichler P., Volz SE., Joung J., van der Oost J., Regev A., Koonin EV., Zhang F. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015 Oct 22;163(3):759-71.
- Arimbasseri AG., Rijal K., Maraia RJ. Transcription termination by the eukaryotic RNA polymerase III. Biochim Biophys Acta. 2013 Mar-Apr;1829(3-4):318-30.
- Rath D., Amlinger L., Rath A., Lundgren M. The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie. 2015 Oct;117:119-28.